Fare login con il vostro nome e password. Uscire con logout o exit e rientrare.
Esaminare il contenuto della vostra home directory con i comandi ls, ls -a, ls -l, ls -la; usare man ls per capire il significato di queste opzioni del comando (per uscire da man battere q).
I comandi per muoversi tra le directory sono pwd e cd. Usare whatis (ed eventualmente man ) per ripassare a cosa serve ciascuno di questi comandi. Visualizzare il nome della propria home directory. Portarsi sulla directory /etc/sysconfig e visualizzarne il contenuto. Quante sotto-directory ci sono?
Spesso, in UNIX, un comando agisce diversamente a seconda dei suoi argomenti. Ad es: ls seguito da uno o più nomi di file visualizza l'elenco relativo a questi file; seguito dal nome di una directory visualizza l'elenco dei file contenuti in questa directory. Verificarlo, facendo una lista lunga dei soli file samba e soundcard, e della directory networking, prima separatamente e poi con un solo comando. Dare il comando "ls s*": potete capire cosa vuol dire il carattere * ? Dare il comando "ls network*" e spiegare quello che viene visualizzato.
Riportarsi sulla propria home directory. Usando pwd e cd, verificare che cosa sono le directory ~ e ..
Qual è il comando per creare una directory: md, mkdir o mdir? Creare la directory tmp nella propria home directory. Come si può indicare il nome di questa directory? Che differenza c'è tra /tmp e ~/tmp?
I principali comandi per agire sui file sono cp, mv, rm. Cosa fa ciascuno? Notare il comportamento diverso di cp e mv a seconda che siano seguiti da due o da più argomenti. Copiare tutti i file della directory /etc/sysconfig che cominciano per "d" nella directory ~/tmp appena creata
Il comando cat visualizza il contenuto di un file. Usarlo per visualizzare il contenuto di uno dei file nelle vostre directory.
Il comando finger visualizza gli utenti collegati sul vostro computer. Usare il carattere di riderezione dell'output (>) per mettere l'output di questo comando nel file ut.collegati.
Usare il comando wc < ut.collegati per stampare su output il numero di righe, parole e caratteri di questo file.
Ottenere lo stesso risultato combinando i comandi dei due
esercizi precedenti con la pipeline
finger | wc
Usando il comando echo, scrivere il vostro nome e cognome sul file mio.cog e il vostro indirizzo (se volete, anche inventato) sul file mio.ind
Sempre usando echo, aggiungere in coda a mio.ind il vostro indirizzo di posta elettronica.
Anche cat può avere due o più argomenti. Usarlo per riversare il contenuto di mio.cog e mio.ind nel file ~/.plan. Usare il carattere di troncamento (*) per riferirsi ai due file con un'unica espressione.
* editare, con vi, il file note. Scrivere in questo file qualche frase di osservazioni e commenti (seri) sul corso.
Usare il comando yppasswd per cambiare la vostra password (lettere minuscole e numeri).
* fare la lista LUNGA dei file sulla vostra home directory, COMPRESI I FILE NASCOSTI. Mettere questa lista, il file .plan e il file note in un unico file e inviarlo per posta elettronica dandolo come input del comando "mail giorgio.signorini@unifi.it"
Fare il numero maggiore possibile di esercizi, nell'ordine. Prima della fine fare comunque l'ultimo esercizio. Gli esercizi contrassegnati con * danno punteggio per la valutazione
* Rendere la vostra home directory leggibile ed eseguibile dal gruppo
* Creare la directory ~/ic-xxxxxx, sostituendo a xxxxxx il nome del vostro utente, che si trova nella variabile USER (esempio: ~/ic-rossi). D'ora in avanti chiameremo questa directory la vostra directory di lavoro
Le variabili HOSTNAME e TERM contengono, rispettivamente, il nome del vostro calcolatore e il tipo di terminale. Stampare su video il loro contenuto. Stampare su video la frase "Benvenuti su ...; il terminale sembra di tipo ..." sostituendo a ... il nome del vostro calcolatore e il tipo di terminale. NB: il carattere ";" deve risultare stampato su video
* Il file ~/.bash_profile contiene comandi da eseguire ad ogni login. Con l'editor vi, editare questo file e inserirvi (come ultima riga) il comando che stampa la frase dell'esercizio precedente. Copiare ~/.bash_profile sulla vostra directory di lavoro
* Fare una lista (lunga) di tutti i file sulla directory /etc il cui nome comincia con "pr" e finisce con "e" oppure "l" (lettera "elle"). Mettere il risultato nel file "pre" sulla vostra directory di lavoro.
Lanciare da terminale il programma netscape. Interromperlo defitivamente battendo ^C (Ctrl C) sullo stesso terminale (non chiudere la finestra di netscape con il mouse!). Rilanciare netscape in background. Visitare la pagina http://NS1.SM.CHIM.UNIFI.IT/~signo/did/index.html
Con il comando jobs fare la lista dei processi in background. Con il comando "kill -9 %<numerojob>" arrestare (definitivamente) il processo di netscape.
* Il comando "ypcat passwd" manda in output le informazioni relative a tutti gli utenti del sistema, un utente per riga (il nome dello user è la serie di caratteri fino al primo ":"; il nome anagrafico dell'utente è la serie di caratteri tra il quarto e il quinto ":"). Usare questo comando in combinazione con il comando wc per stabilire quanti sono gli utenti del sistema. Scrivere (con vi) la risposta nel file utenti nella vostra directory di lavoro
* Usare "ypcat passwd" in combinazione con il comando grep per stampare su video le righe relative a tutti gli utenti che si chiamano (anagraficamente) "Francesco" o "Francesca". Dirigere l'output nel file "Francesc" sulla vostra directory di lavoro.
* Ripetere l'esercizio precedente, includendo tutte le righe in cui "Francesco" o "Francesca" sono scritti sia con l'iniziale maiuscola che con la minuscola. Dirigere l'output nel file "francesc" sulla vostra directory di lavoro.
* Usando il comando sort, mettere in ordine alfabetico di user il file "francesc".Dirigere l'output nel file "francesc.sorted" sulla vostra directory di lavoro.
* Rendere il file ~/.bash_history leggibile dal gruppo. Uscire dal terminale con exit. Riaprire un terminale e copiare il file ~/.bash_history sulla vostra directory di lavoro.
Il dipolo di una molecola dove siano definite delle cariche atomiche è una quantità vettoriale data dalla formula
![]()
dove l'indice i scorre sugli atomi, qi è la carica dell'atomo i, e ri è la sua posizione.
Scrivere un programma in awk (~/ese/dipolo.awk) per calcolare il dipolo della molecola XXX usando le coordinate atomiche contenute nel file ~infochim/e3/xxx.str e le cariche atomiche che sono riportate nella prima riga del file ~infochim/e3/xxx.chg.
N.B. A ciascuno viene assegnata una molecola XXX !
Ripetere il calcolo usando volta a volta le serie di cariche atomiche elencate in ogni riga del file xxx.chg. Riportare i risultati in una tabella (file ~/ese/dipolo.tab) che per ogni serie di cariche contenga una riga e cinque colonne: carica del primo atomo, dipolo (componenti x, y e z e valore assoluto).
Creare la directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro); portarsi sulla directory di lavoro e copiarvi i seguenti file contenuti nella directory ~infochim/wrk/e3:
xxx.str
tutti i file il cui nome finisce in .awk
Il file xxx.str contiene le coordinate degli atomi della molecola XXX, un atomo per riga, con il seguente formato:
(specie atomica) xi yi zi
Esaminare il contenuto di questo file usando i comando cat o il programma vi.
Il file r.awk
contiene un programma in awk che eseguito sul file xxx.str
stampa su standard output gli stessi dati seguiti dal modulo del
vettore posizione dell'atomo![]() :
:
(specie atomica) xi yi zi ri
...
Lanciare un comando awk che esegua questo programma sul file di dati xxx.str. Dirigere il risultato prima su standard output (terminale), poi sul file xxx.r
modificare il programma r.awk (editarlo con vi) in modo tale che vengano stampati solo i dati relativi al secondo e terzo atomo che compaiono in xxx.str. Eseguire questo nuovo programma sul file di dati; dirigere il risultato prima su standard output, poi sul file xxx.a
scrivere un programma awk (aprire con vi un nuovo file dipolo.awk)che calcoli il momento di dipolo della molecola a partire da cariche atomiche date. Il momento di dipolo è dato dalla seguente formula:
![]()
dove l'indice i scorre sugli atomi, qi è la carica dell'atomo i, e ri è la sua posizione..
Il programma deve scrivere su output le tre componenti del dipolo:
dx dy dz
I valori di carica atomica da utilizzare sono i seguenti:
qyyy = ...
qzzz = ...
Suggerimento: via via che il programma legge una riga del file di input con i dati relativi ad un atomo, fargli calcolare il contributo di questo atomo alle tre componenti di d: qi xi, qi yi, e qi zi, sommandolo ai contributi degli atomi precedenti.
Dopo avere processato tutte le righe, far stampare la somma di tutti contributi.
Eseguire dipolo.awk sul file di dati; dirigere il risultato su xxx.d
Modificare il programma dipolo.awk
in modo tale che alla fine stampi, oltre a dx, dy,
dz, anche il modulo
del dipolo:![]()
Eseguire il programma sul file di dati e dirigere il risultato sul file xxx.dip.
Creare la directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro); portarsi sulla directory di lavoro e copiarvi i seguenti file contenuti nella directory ~infochim/wrk/e3:
ncl2.str
tutti i file il cui nome finisce in .awk
Il file ncl2.str contiene le coordinate degli atomi della molecola NCl2, un atomo per riga, con il seguente formato:
(specie atomica) xi yi zi
Esaminare il contenuto di questo file usando i comando cat o il programma vi.
Il file r.awk
contiene un programma in awk che eseguito sul file ncl2.str
stampa su standard output gli stessi dati seguiti dal modulo del
vettore posizione dell'atomo![]() :
:
(specie atomica) xi yi zi ri
...
Lanciare un comando awk che esegua questo programma sul file di dati ncl2.str. Dirigere il risultato prima su standard output (terminale), poi sul file ncl2.r
modificare il programma r.awk (editarlo con vi) in modo tale che vengano stampati solo i dati relativi al secondo e terzo atomo che compaiono in ncl2.str. Eseguire questo nuovo programma sul file di dati; dirigere il risultato prima su standard output, poi sul file ncl2.a
scrivere un programma awk (aprire con vi un nuovo file dipolo.awk)che calcoli il momento di dipolo della molecola a partire da cariche atomiche date. Il momento di dipolo è dato dalla seguente formula:
![]()
dove l'indice i scorre sugli atomi, qi è la carica dell'atomo i, e ri è la sua posizione..
Il programma deve scrivere su output le tre componenti del dipolo:
dx dy dz
I valori di carica atomica da utilizzare sono i seguenti:
qN = -0.176
qCl = 0.088
Suggerimento: via via che il programma legge una riga del file di input con i dati relativi ad un atomo, fargli calcolare il contributo di questo atomo alle tre componenti di d: qi xi, qi yi, e qi zi, sommandolo ai contributi degli atomi precedenti.
Dopo avere processato tutte le righe, far stampare la somma di tutti contributi.
Eseguire dipolo.awk sul file di dati; dirigere il risultato su ncl2.d
Modificare il programma dipolo.awk
in modo tale che alla fine stampi, oltre a dx, dy,
dz, anche il modulo
del dipolo:![]()
Eseguire il programma sul file di dati e dirigere il risultato sul file ncl2.dip.
Creare la directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro); portarsi sulla directory di lavoro e copiarvi i seguenti file contenuti nella directory ~infochim/wrk/e3:
no2.str
tutti i file il cui nome finisce in .awk
Il file no2.str contiene le coordinate degli atomi della molecola NO2, un atomo per riga, con il seguente formato:
(specie atomica) xi yi zi
Esaminare il contenuto di questo file usando i comando cat o il programma vi.
Il file r.awk
contiene un programma in awk che eseguito sul file no2.str
stampa su standard output gli stessi dati seguiti dal modulo del
vettore posizione dell'atomo![]() :
:
(specie atomica) xi yi zi ri
...
Lanciare un comando awk che esegua questo programma sul file di dati no2.str. Dirigere il risultato prima su standard output (terminale), poi sul file no2.r
Modificare il programma r.awk (editarlo con vi) in modo tale che vengano stampati solo i dati relativi al secondo e terzo atomo che compaiono in no2.str. Eseguire questo nuovo programma sul file di dati; dirigere il risultato prima su standard output, poi sul file no2.a
scrivere un programma awk (aprire con vi un nuovo file dipolo.awk)che calcoli il momento di dipolo della molecola a partire da cariche atomiche date. Il momento di dipolo è dato dalla seguente formula:
![]()
dove l'indice i scorre sugli atomi, qi è la carica dell'atomo i, e ri è la sua posizione..
Il programma deve scrivere su output le tre componenti del dipolo:
dx dy dz
I valori di carica atomica da utilizzare sono i seguenti:
qN = 0.214
qO = -0.107
Suggerimento: via via che il programma legge una riga del file di input con i dati relativi ad un atomo, fargli calcolare il contributo di questo atomo alle tre componenti di d: qi xi, qi yi, e qi zi, sommandolo ai contributi degli atomi precedenti.
Dopo avere processato tutte le righe, far stampare la somma di tutti contributi.
Eseguire dipolo.awk sul file di dati; dirigere il risultato su no2.d
Modificare il programma dipolo.awk
in modo tale che alla fine stampi, oltre a dx, dy,
dz, anche il modulo
del dipolo:![]()
Eseguire il programma sul file di dati e dirigere il risultato sul file no2.dip.
1. Creare la directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro); portarsi sulla directory di lavoro e copiarvi i seguenti file contenuti nella directory ~infochim/wrk/e3:
xxx.str
tutti i file il cui nome finisce in .awk
2. Il file xxx.str contiene le coordinate degli atomi della molecola XXX, un atomo per riga, con il seguente formato:
(specie atomica) xi yi zi
Esaminare il contenuto di questo file usando i comando cat o il programma vi.
Il file r.awk
contiene un programma in awk che eseguito sul file xxx.str
stampa su standard output gli stessi dati seguiti dal modulo del
vettore posizione dell'atomo![]() :
:
(specie atomica) xi yi zi ri
...
Lanciare un comando awk che esegua questo programma sul file di dati xxx.str. Dirigere il risultato prima su standard output (terminale), poi sul file xxx.r
3. Modificare il programma r.awk (editarlo con vi) in modo tale che vengano stampati solo i dati relativi al secondo e terzo atomo che compaiono in xxx.str. Eseguire questo nuovo programma sul file di dati; dirigere il risultato prima su standard output, poi sul file xxx.a
4. Scrivere un programma awk (aprire con vi un nuovo file cmassa.awk)che calcoli la posizione del centro di massa della molecola, R:
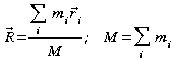
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e ri è la sua posizione.Ovvero:
![]()
Il programma deve scrivere su output le tre componenti di R:
Rx Ry Rz
Le masse atomiche sono: mH = 1.008; mO = 15.999; mN = 14.007;
Suggerimento: via via che il programma legge una riga del file di input con i dati relativi ad un atomo, fargli calcolare il contributo di questo atomo a M, mi , e alle tre componenti di R: mi xi, mi yi, e mi zi, sommando questi contributi ai valori di M, Rx,Ry,Rz già accumulati.
Dopo avere processato tutte le righe, far stampare la somma di tutti contributi (m1 x1+m2 x2+...)/M; etc.
Eseguire cmassa.awk sul file di dati; dirigere il risultato su no2.cm
5. Modificare il programma cmassa.awk
in modo tale che alla fine stampi, oltre a Rx, Ry,
Rz, anche il modulo
di R:![]()
Eseguire il programma sul file di dati e dirigere il risultato sul file no2.cmass.
1. Creare la directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro); portarsi sulla directory di lavoro e copiarvi i seguenti file contenuti nella directory ~infochim/wrk/e3:
h2o.str
tutti i file il cui nome finisce in .awk
2. Il file h2o.str contiene le coordinate degli atomi della molecola H2O, un atomo per riga, con il seguente formato:
(specie atomica) xi yi zi
Esaminare il contenuto di questo file usando i comando cat o il programma vi.
Il file r.awk
contiene un programma in awk che eseguito sul file h2o.str
stampa su standard output gli stessi dati seguiti dal modulo del
vettore posizione dell'atomo![]() :
:
(specie atomica) xi yi zi ri
...
Lanciare un comando awk che esegua questo programma sul file di dati h2o.str. Dirigere il risultato prima su standard output (terminale), poi sul file h2o.r
3. Modificare il programma r.awk (editarlo con vi) in modo tale che vengano stampati solo i dati relativi al secondo e terzo atomo che compaiono in h2o.str. Eseguire questo nuovo programma sul file di dati; dirigere il risultato prima su standard output, poi sul file h2o.a
4. Scrivere un programma awk (aprire con vi un nuovo file cmassa.awk)che calcoli la posizione del centro di massa della molecola, R:
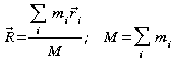
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e ri è la sua posizione.Ovvero:
![]()
Il programma deve scrivere su output le tre componenti di R:
Rx Ry Rz
Le masse atomiche sono: mH = 1.008; mO = 15.999; mN = 14.007;
Suggerimento: via via che il programma legge una riga del file di input con i dati relativi ad un atomo, fargli calcolare il contributo di questo atomo a M, mi , e alle tre componenti di R: mi xi, mi yi, e mi zi, sommando questi contributi ai valori di M, Rx,Ry,Rz già accumulati.
Dopo avere processato tutte le righe, far stampare la somma di tutti contributi (m1 x1+m2 x2+...)/M; etc.
Eseguire cmassa.awk sul file di dati; dirigere il risultato su no2.cm
5. Modificare il programma cmassa.awk
in modo tale che alla fine stampi, oltre a Rx, Ry,
Rz, anche il modulo
di R:![]()
Eseguire il programma sul file di dati e dirigere il risultato sul file no2.cmass.
1. Creare la directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro); portarsi sulla directory di lavoro e copiarvi i seguenti file contenuti nella directory ~infochim/wrk/e3:
no2.str
tutti i file il cui nome finisce in .awk
2. Il file no2.str contiene le coordinate degli atomi della molecola NO2, un atomo per riga, con il seguente formato:
(specie atomica) xi yi zi
Esaminare il contenuto di questo file usando i comando cat o il programma vi.
Il file r.awk
contiene un programma in awk che eseguito sul file no2.str
stampa su standard output gli stessi dati seguiti dal modulo del
vettore posizione dell'atomo![]() :
:
(specie atomica) xi yi zi ri
...
Lanciare un comando awk che esegua questo programma sul file di dati no2.str. Dirigere il risultato prima su standard output (terminale), poi sul file no2.r
3. Modificare il programma r.awk (editarlo con vi) in modo tale che vengano stampati solo i dati relativi al secondo e terzo atomo che compaiono in no2.str. Eseguire questo nuovo programma sul file di dati; dirigere il risultato prima su standard output, poi sul file no2.a
4. Scrivere un programma awk (aprire con vi un nuovo file cmassa.awk)che calcoli la posizione del centro di massa della molecola, R:
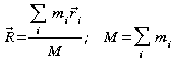
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e ri è la sua posizione.Ovvero:
![]()
Il programma deve scrivere su output le tre componenti di R:
Rx Ry Rz
Le masse atomiche sono: mH = 1.008; mO = 15.999; mN = 14.007;
Suggerimento: via via che il programma legge una riga del file di input con i dati relativi ad un atomo, fargli calcolare il contributo di questo atomo a M, mi , e alle tre componenti di R: mi xi, mi yi, e mi zi, sommando questi contributi ai valori di M, Rx,Ry,Rz già accumulati.
Dopo avere processato tutte le righe, far stampare la somma di tutti contributi (m1 x1+m2 x2+...)/M; etc.
Eseguire cmassa.awk sul file di dati; dirigere il risultato su no2.cm
5. Modificare il programma cmassa.awk
in modo tale che alla fine stampi, oltre a Rx, Ry,
Rz, anche il modulo
di R:![]()
Eseguire il programma sul file di dati e dirigere il risultato sul file no2.cmass.
1. Portarsi sulla directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro) e copiarvi, dalla directory ~infochim/wrk/e4 , il file 12diazetidine.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominiciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
Le righe che cominicano con un'altra parola contengono altri dati sulla molecola.
2. Scrivere un programma awk (aprire con vi un nuovo file inerzia.awk)che calcoli il momento di inerzia della molecola intorno all'asse y:
![]()
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e xi , zi sono le sue coordinate x e z.
Ovvero:
![]()
Il programma deve scrivere su output, in un solo rigo e senza
stringhe di commento, due numeri: la massa totale
![]() e
e![]()
Dirigere l'output prima su terminale, poi sul file inerzia.tab
Le masse atomiche sono: mH = 1.008; mC = 12.011; mN = 14.007; mO = 15.999;
3. Ripetere il calcolo modificando a turno la massa di un atomo con le masse seguenti (che corrispondono a isotopi naturali):
mH = 2.014;
mC = 13.003;
mN = 15.000;
mO = 16.999; 17.999
dirigere l'output prima su terminale, poi appenderlo in coda a inerzia.tab
4. Usando gnuplot, diagrammare i dati contenuti in inerzia.tab : momento di inerzia contro massa totale.
Dopo
avere visualizzato il grafico su schermo, salvarlo in formato
PostScript nel file inerzia.ps
dando il seguente comando:
set term postscript; set out 'inerzia.ps'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out
1. Portarsi sulla directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro) e copiarvi, dalla directory ~infochim/wrk/e4 , il file acetimidine.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominiciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
Le altre righe contengono altri dati sulla molecola.
2. Scrivere un programma awk (aprire con vi un nuovo file inerzia.awk)che calcoli il momento di inerzia della molecola intorno all'asse y:
![]()
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e xi , zi sono le sue coordinate x e z.
Ovvero:
![]()
Il programma deve scrivere su output, in un solo rigo e senza
stringhe di commento, due numeri: la massa totale
![]() e
e![]()
Dirigere l'output prima su terminale, poi sul file inerzia.tab
Le masse atomiche sono: mH = 1.008; mC = 12.011; mN = 14.007; mO = 15.999;
3. Ripetere il calcolo modificando a turno la massa di un atomo con le masse seguenti (che corrispondono a isotopi naturali):
mH = 2.014;
mC = 13.003;
mN = 15.000;
mO = 16.999; 17.999
dirigere l'output prima su terminale, poi appenderlo in coda a inerzia.tab
4.
Usando gnuplot, diagrammare i dati contenuti in
inerzia.tab:: momento
di inerzia contro massa totale. Salvare il grafico in formato JPEG
nel file inerzia.jpeg.
dando il seguente comando:
set term jpeg; set out 'inerzia.jpeg'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out
1. Portarsi sulla directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro) e copiarvi, dalla directory ~infochim/wrk/e4 , il file glycerald.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
(Le righe che cominiciano con un'altra parola contengono altri dati sulla molecola).
Scrivere un programma awk (aprire con vi un nuovo file coord.awk)che stampi solo le righe con le coordinate dell'idrogeno.
2. Scrivere un programma awk (aprire con vi un nuovo file inerzia.awk)che calcoli il momento di inerzia della molecola intorno all'asse z:
![]()
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e xi , yi sono le sue coordinate x e y.
Ovvero:
![]()
Il programma deve scrivere su output, in un solo rigo e senza
stringhe di commento, due numeri: la massa totale
![]() e
e![]()
Dirigere l'output prima su terminale, poi sul file inerzia.tab
Le masse atomiche sono: mH = 1.008; mC = 12.011; mN = 14.007; mO = 15.999;
3. Ripetere il calcolo modificando a turno la massa di un atomo con le masse seguenti (che corrispondono a isotopi naturali):
mH = 2.014;
mC = 13.003;
mN = 15.000;
mO = 16.999; 17.999
dirigere l'output prima su terminale, poi appenderlo in coda a inerzia.tab
4. Usando gnuplot, diagrammare i dati contenuti in inerzia.tab : momento di inerzia contro massa totale.
Dopo
avere visualizzato il grafico su schermo, salvarlo in formato
PostScript nel file inerzia.ps
dando il seguente comando:
set term postscript; set out 'inerzia.ps'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out
1. Portarsi sulla directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro) e copiarvi, dalla directory ~infochim/wrk/e4 , il file lactacid.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
(Le righe che cominiciano con un'altra parola contengono altri dati sulla molecola).
Scrivere un programma awk (aprire con vi un nuovo file coord.awk)che stampi solo le righe con le coordinate dell'idrogeno.
2. Scrivere un programma awk (aprire con vi un nuovo file inerzia.awk)che calcoli il momento di inerzia della molecola intorno all'asse x:
![]()
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e yi , zi sono le sue coordinate y e z.
Ovvero:
![]()
Il programma deve scrivere su output, in un solo rigo e senza
stringhe di commento, due numeri: la massa totale
![]() e
e![]()
Dirigere l'output prima su terminale, poi sul file inerzia.tab
Le masse atomiche sono: mH = 1.008; mC = 12.011; mN = 14.007; mO = 15.999;
3. Ripetere il calcolo modificando a turno la massa di un atomo con le masse seguenti (che corrispondono a isotopi naturali):
mH = 2.014;
mC = 13.003;
mN = 15.000;
mO = 16.999; 17.999
dirigere l'output prima su terminale, poi appenderlo in coda a inerzia.tab
4. Usando gnuplot, diagrammare i dati contenuti in inerzia.tab : momento di inerzia contro massa totale.
Dopo
avere visualizzato il grafico su schermo, salvarlo in formato
PostScript nel file inerzia.ps
dando il seguente comando:
set term postscript; set out 'inerzia.ps'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out
1. Portarsi sulla directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro) e copiarvi, dalla directory ~infochim/wrk/e4 , il file glycerald.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
(Le righe che cominiciano con un'altra parola contengono altri dati sulla molecola).
Scrivere un programma awk (aprire con vi un nuovo file coord.awk)che stampi solo le righe con le coordinate dell'idrogeno.
2. Scrivere un programma awk (aprire con vi un nuovo file inerzia.awk)che calcoli il momento di inerzia della molecola intorno all'asse x:
![]()
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e yi , zi sono le sue coordinate y e z.
Ovvero:
![]()
Il programma deve scrivere su output, in un solo rigo e senza
stringhe di commento, due numeri: la massa totale
![]() e
e![]()
Dirigere l'output prima su terminale, poi sul file inerzia.tab
Le masse atomiche sono: mH = 1.008; mC = 12.011; mN = 14.007; mO = 15.999;
3. Ripetere il calcolo modificando a turno la massa di un atomo con le masse seguenti (che corrispondono a isotopi naturali):
mH = 2.014;
mC = 13.003;
mN = 15.000;
mO = 16.999; 17.999
dirigere l'output prima su terminale, poi appenderlo in coda a inerzia.tab
4. Usando gnuplot, diagrammare i dati contenuti in inerzia.tab : momento di inerzia contro massa totale.
Dopo
avere visualizzato il grafico su schermo, salvarlo in formato
PostScript nel file inerzia.ps
dando il seguente comando:
set term postscript; set out 'inerzia.ps'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out
1. Portarsi sulla directory ~/ese (d'ora in poi la chiameremo la vostra directory di lavoro) e copiarvi, dalla directory ~infochim/wrk/e4 , il file lactacid.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
(Le righe che cominiciano con un'altra parola contengono altri dati sulla molecola).
Scrivere un programma awk (aprire con vi un nuovo file coord.awk)che stampi solo le righe con le coordinate dell'idrogeno.
2. Scrivere un programma awk (aprire con vi un nuovo file inerzia.awk)che calcoli il momento di inerzia della molecola intorno all'asse z:
![]()
dove l'indice i scorre sugli atomi, mi è la massa dell'atomo i, e xi , yi sono le sue coordinate x e y.
Ovvero:
![]()
Il programma deve scrivere su output, in un solo rigo e senza
stringhe di commento, due numeri: la massa totale
![]() e
e![]()
Dirigere l'output prima su terminale, poi sul file inerzia.tab
Le masse atomiche sono: mH = 1.008; mC = 12.011; mN = 14.007; mO = 15.999;
3. Ripetere il calcolo modificando a turno la massa di un atomo con le masse seguenti (che corrispondono a isotopi naturali):
mH = 2.014;
mC = 13.003;
mN = 15.000;
mO = 16.999; 17.999
dirigere l'output prima su terminale, poi appenderlo in coda a inerzia.tab
4. Usando gnuplot, diagrammare i dati contenuti in inerzia.tab : momento di inerzia contro massa totale.
Dopo
avere visualizzato il grafico su schermo, salvarlo in formato
PostScript nel file inerzia.ps
dando il seguente comando:
set term postscript; set out 'inerzia.ps'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out
1. Portarsi sulla directory ~/ese e copiarvi, dalla directory ~infochim/wrk/e4 , il file dmso.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
(Le righe che cominiciano con un'altra parola contengono altri dati sulla molecola).
Scrivere un programma awk (aprire con vi un nuovo file coord.awk)che stampi solo le righe con le coordinate dell'idrogeno.
2. Scrivere un programma awk
(aprire con vi un nuovo file dip.awk)che
calcoli il momento di dipolo della molecola utilizzando le coordinate
del file dell'esercizio precedente e le cariche atomiche riportate
qui sotto.
Il momento di dipolo è dato dalla seguente
formula:
![]()
dove l'indice i scorre sugli atomi, qi è la carica dell'atomo i, e ri è la sua posizione.. Ovvero:
![]()
I valori di carica atomica da utilizzare sono i seguenti:
qS =0.5; qO =-0.5; qC =0.45; qH =-0.15;
Il programma deve scrivere su output in
un solo rigo e senza stringhe di commento, due numeri: la
carica sull'ossigeno qO e il modulo del dipolo:![]()
Eseguire dipolo.awk sul file di dati; dirigere il risultato prima su terminale, poi su dip.tab
3. Ripetere il calcolo utilizzando volta a volta, al posto dell'insieme di cariche dell'esercizio precedente, uno dei seguenti insiemi:
qS =0.139; qO =-0.459; qC =0.16; qH =0;
qS =0.139; qO =-0.439; qC =-0.29; qH =-0.15;
qS =0.64; qO =-0.55; qC =-0.645; qH =-0.2;
dirigere l'output prima su terminale, poi appenderlo in coda a dip.tab
4.
Usando gnuplot, diagrammare i dati contenuti in dip.tab
: carica dell'ossigeno contro modulo del dipolo. Dopo
avere visualizzato il grafico su schermo, salvarlo in formato
PostScript nel file dip.ps
dando il seguente comando:
set term postscript; set out 'dip.ps'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out
1. Portarsi sulla directory ~/ese e copiarvi, dalla directory ~infochim/wrk/e4 , il file dmether.pdb
Questo file contiene le coordinate di una molecola scritte nel formato PDB: le coordinate si trovano nelle righe che cominciano per HETATM, una riga per atomo; la specie atomica e le coordinate x, y e z sono nelle posizioni sottolineate nel seguente esempio:
HETATM 2 C 1 1.548 0.000 0.000 1.00 0.00
(Le righe che cominiciano con un'altra parola contengono altri dati sulla molecola).
Scrivere un programma awk (aprire con vi un nuovo file coord.awk)che stampi solo le righe con le coordinate dell'idrogeno.
2. Scrivere un programma awk
(aprire con vi un nuovo file dip.awk)che
calcoli il momento di dipolo della molecola utilizzando le coordinate
del file dell'esercizio precedente e le cariche atomiche riportate
qui sotto.
Il momento di dipolo è dato dalla seguente
formula:
![]()
dove l'indice i scorre sugli atomi, qi è la carica dell'atomo i, e ri è la sua posizione.. Ovvero:
![]()
I valori di carica atomica da utilizzare sono i seguenti:
qS =0.5; qO =-0.5; qC =0.45; qH =-0.15;
Il programma deve scrivere su output in
un solo rigo e senza stringhe di commento, due numeri: la
carica sull'ossigeno qO e il modulo del dipolo:![]()
Eseguire dipolo.awk sul file di dati; dirigere il risultato prima su terminale, poi su dip.tab
3. Ripetere il calcolo utilizzando volta a volta, al posto dell'insieme di cariche dell'esercizio precedente, uno dei seguenti insiemi:
qS =0.139; qO =-0.459; qC =0.16; qH =0;
qS =0.139; qO =-0.439; qC =-0.29; qH =-0.15;
qS =0.64; qO =-0.55; qC =-0.645; qH =-0.2;
dirigere l'output prima su terminale, poi appenderlo in coda a dip.tab
4.
Usando gnuplot, diagrammare i dati contenuti in dip.tab
: carica dell'ossigeno contro modulo del dipolo. Dopo
avere visualizzato il grafico su schermo, salvarlo in formato
PostScript nel file dip.ps
dando il seguente comando:
set term postscript; set out 'dip.ps'
ripetendo poi il comando plot ... che genera il grafico.
NB:
Per tornare a mostrare il grafico sullo schermo, dare il comando:
set term X11; set out